IVDR CE认证是欧盟最新的法规要求!
IVDR根据2017/746 法规第 V 章对IVDR医疗器械的符合性评估程序所做要求,基于产品的风险将所有的体外诊断设备由低到高分为了A、B、C、D四类。
根据法规要求,A(无菌)、 B、C和D类产品需要公告机构审核发证;A类不需要公告机构的介入,由欧代完成CE备案,如选择IVDR机构作为欧代,IVDR机构帮您完成A类CE备案。
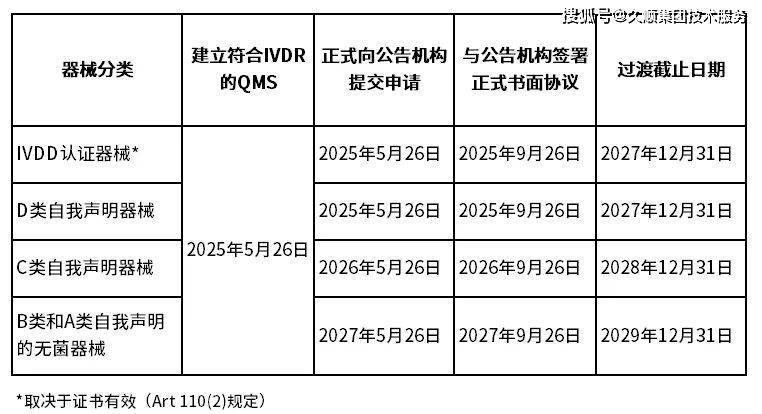
2024年欧盟发布IVDD延期声明,声明中,不同分类的IVD器械可适用新的过渡截止期限,详见下表:
1.符合98/79/EC指令,并且不会对患者、使用者、其他人员的健康或安全,或对公共健康保护构成不可接受的风险。
2.器械的设计或预期用途未发生重大变化。
3.制造商需在2025年5月26日前,建立符合IVDR法规的质量管理体系(QMS)。
4.制造商需在下述适用的截止日期前提交 IVDR申请,并与公告机构签署正式书面协议。
目前,IVD企业有两个选择,一是直接申请IVDR CE认证;二是在拥有IVDD CE证书前提下,选择签署延期协议,申请IVDD CE延期。
无论做哪个选择选择,企业都需要做:
1.在2025年5月26日前,建立符合IVDR法规的质量管理体系(QMS);
2.编写IVDR CE技术文档;
3.和认证机构签署协议;
以上的工作内容,如您需要找专业的合作伙伴指引办证方向,IVDR机构 作为专业的IVD咨询公司,可以找IVDR机构进行咨询,IVDR机构是IVD企业重要的合作伙伴。